 |
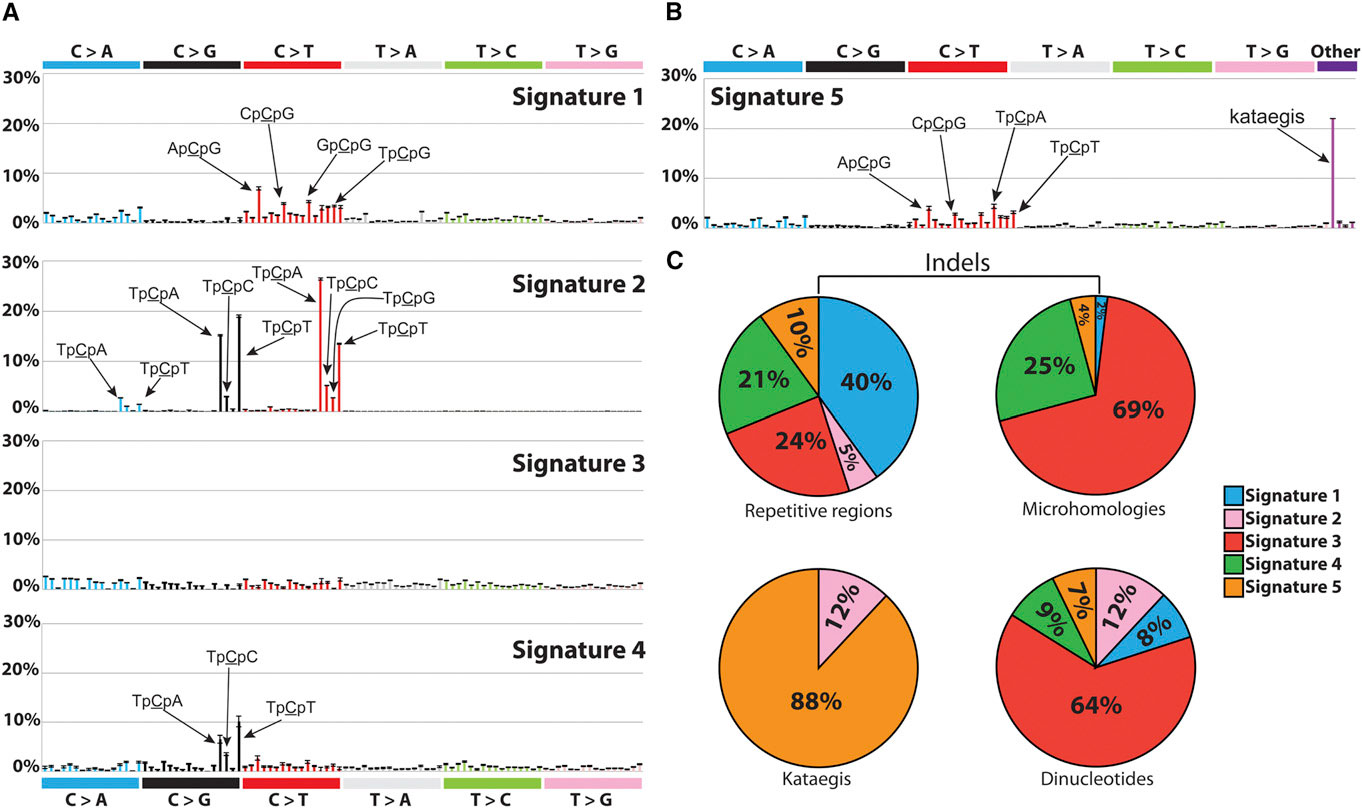
| Signatures of Mutational Processes Extracted from the Mutational Catalogs of 21 Breast Cancer Genomes. Credit:http://dx.doi.org/10.1016/j.celrep.2012.12.008 |
Cancer is the second leading cause of death worldwide, with approximately 14 million new cases and 8.2 million cancer related deaths each year (Source: WHO). A family history of cancer typically increases a person's risk of developing the disease, yet most cancer cases have no family history at all. This suggests that a combination of both genetics and environmental exposures contribute to the etiology of cancer. In this context, "genetics" means the genetic make-up we are born with and inherited from our parents. For example, women born with specific mutations in the BRCA1 and BRCA2 genes are known to have a much higher risk of developing breast cancer later in life.
However, besides the genetic make-up we carry from birth, there are many geographical and environmental factors that contribute to the risk of cancer. For example, the incidence of breast cancer is over 4 times higher in North and West Europe compared to Asia and Africa (Source: WHO). Stomach cancer, on the other hand, is much more prevalent in Asia than the US. If you think that this may be linked to the genetic differences across ethnicities, think again. The National Cancer Institute published a summary of several studies that compared the incidence of first and second generation immigrants in the US with the local population. They found that:
"cancer incidence patterns among first-generation immigrants were nearly identical to those of their native country, but through subsequent generations, these patterns evolved to resemble those found in the United States. This was true especially for cancers related to hormones, such as breast, prostate, and ovarian cancer and neoplasms of the uterine corpus and cancers attributable to westernized diets, such as colorectal malignancies."According to the World Health Organization (WHO),
"around one third of cancer deaths are due to the 5 leading behavioral and dietary risks: high body mass index, low fruit and vegetable intake, lack of physical activity, tobacco use, alcohol use."Cancer is the result of a series of cellular mechanisms gone awry: every time a cell divides, somatic mutations accumulate in the cell's genome. These are not mutations we are born with, inherited from our parents. Rather, these are changes that accumulate in certain cells as we grow old and are not the same across all cells in the body. Many environmental exposures contribute to this process and affect the rate at which these mutations accumulate. However, cells have various mechanisms that are normally able to repair harmful mutations or, when the damage is beyond repair, to trigger cell death. The immune system is also "trained" to recognize cancer cells and destroy them.
When all these defense mechanisms fail, cancer cells start dividing uncontrollably.
As a result, all cancer cells carry a number of somatic mutations that set them apart from healthy cells, and some tend to be the same across different cancer patients: for example, specific mutational patterns found in lung cancer have been attributed to tobacco exposure and were indeed reproduced in animal models. Another set of mutations has been attributed to UV exposure and has been found in skin cancers [1, 2].
This prompts the ambitious question: can we find common mutations across individuals with the same cancer? And how many of these mutational patterns that are common across individuals can we attribute to particular exposures and/or biological processes? Distinguished postdoctoral researcher Ludmil Alexandrov, from the Los Alamos National Laboratory, has been working on this problem since his he was a PhD student at the Wellcome Trust Sanger Institute.
"It's like lifting fingerprints," Alexandrov explains. "The mutations are the fingerprints, but now we have to do the investigative work and find the 'perpetrator', i.e., the carcinogens that caused them." During his graduate studies, under the supervision of Mike Stratton of the Wellcome Trust Sanger Institute, Alexandrov developed a mathematical model that, given the cancer genomes from a number of patients, is able to pick the "common signals" across the patients -- i.e. mutation patterns that are common across the patients -- and classify them into "signatures."
"When formulated mathematically," Alexandrov explains, "the question can be expressed as the classic 'cocktail party' problem, where multiple people in a room are speaking simultaneously while several microphones placed at different locations are recording the conversations. Each microphone captures a combination of all sounds and the problem is to identify the individual conversations from all the recordings." Taking from this analogy, each cancer genome is a "recording", and the task of the mathematical model is to reconstruct each conversation, in other words, the mutational patterns. These are sets of somatic mutations that are the observed across the cancer genomes and that characterize certain types of cancers.
In 2013, Alexandrov and colleagues analyzed 4,938,362 mutations from 7,042 patients, spanning 30 different cancers, and extracted more than 20 distinct mutational signatures [2]. "Some patterns were expected, like the known ones caused by tobacco and UV light," Alexandrov says. "Others were completely new."
Of the new signatures found, many are involved in defective DNA repair mechanisms, suggesting that drugs targeting these specific mechanisms may benefit cancers exhibiting these signatures [3]. But the most exciting part of this research will be finding the 'perpetrator' or, as Alexandrov explains, the mutations triggered by carcinogens like tobacco, UV radiation, obesity, and so on. The challenge will be to experimentally associate these mutational patterns to the exposures that caused them. In order to do this, the scientists will have to expose cultured cells and model organisms to known carcinogens and then analyze the genomes of the experimentally induced cancers.
In the meantime, the signatures found so far are only the beginning: Alexandrov and colleagues have teamed up with the Los Alamos High Performance Computing Organization in order to analyze the genomes of almost 30,000 cancer patients.
"The amount of data we will have to handle for this task is enormous, on the order of petabytes," Alexandrov says. "Few places in the world have the capability to handle this many data. Under normal circumstances, it takes months to answer a question on 10 petabytes of data. The supercomputing facility at Los Alamos can provide an answer within a day."
Because of his research, in 2014 Alexandrov was listed by Forbes magazine as one of the “30 brightest stars under the age of 30” in the field of Science and Healthcare. In 2015 he was awarded the AAAS Science & SciLifeLab Prize for Young Scientists in the category Genomics and Proteomics [2] and the Weintraub Award for Graduate Research. He is now the recipient of the prestigious Oppenheimer fellowship at Los Alamos National Laboratory.
References
Siegel, R., Miller, K., & Jemal, A. (2015). Cancer statistics, 2015 CA: A Cancer Journal for Clinicians, 65 (1), 5-29 DOI: 10.3322/caac.21254
[1] Alexandrov LB (2015). Understanding the origins of human cancer. Science (New York, N.Y.), 350 (6265) PMID: 26785464
[2] Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, Eils R, Eyfjörd JE, Foekens JA, Greaves M, Hosoda F, Hutter B, Ilicic T, Imbeaud S, Imielinski M, Jäger N, Jones DT, Jones D, Knappskog S, Kool M, Lakhani SR, López-Otín C, Martin S, Munshi NC, Nakamura H, Northcott PA, Pajic M, Papaemmanuil E, Paradiso A, Pearson JV, Puente XS, Raine K, Ramakrishna M, Richardson AL, Richter J, Rosenstiel P, Schlesner M, Schumacher TN, Span PN, Teague JW, Totoki Y, Tutt AN, Valdés-Mas R, van Buuren MM, van 't Veer L, Vincent-Salomon A, Waddell N, Yates LR, Australian Pancreatic Cancer Genome Initiative, ICGC Breast Cancer Consortium, ICGC MMML-Seq Consortium, ICGC PedBrain, Zucman-Rossi J, Futreal PA, McDermott U, Lichter P, Meyerson M, Grimmond SM, Siebert R, Campo E, Shibata T, Pfister SM, Campbell PJ, & Stratton MR (2013). Signatures of mutational processes in human cancer. Nature, 500 (7463), 415-21 PMID: 23945592
[3] Alexandrov LB, Nik-Zainal S, Siu HC, Leung SY, & Stratton MR (2015). A mutational signature in gastric cancer suggests therapeutic strategies. Nature communications, 6 PMID: 26511885